")
")
Оптичният невромиелит (OНМ, neuromyelitis optica), доскоро наричан болест или синдром на Devic, е рядко срещано възпалително, демиелинизиращо, автоимунно В-клетъчно медиирано заболяване на ЦНС [9–11, 30, 31, 33, 35]. Характеризира се с тежко рекурентно увреждане на зрителния нерв, гръбначния мозък и в много по-малка степен на бялото вещество на главния мозък. Това заболяване се различава от множествената склероза (МС) по своята етиология и патогенеза, клинична тежест и терапевтичен подход [1, 2, 9, 11, 14, 25, 31]. Установен е специфичен биологичен маркер – патогенни имуноглобулини, IgG антитела (NMO-IgG) – серологичната диагноза понастоящем е сигурна при 78% от болните, а може да се изключи при чувствителност на тестовете над 90% [13, 14, 18]. Тези антитела са насочени към aquaporin-4 (AQP-4) водни канали, които се експресират в ЦНС [18, 20, 26, 28], в скелетната мускулатура, бъбреците, вътрешното ухо и стомашната мукоза. В над 80% от случаите ОНМ има пристъпно-ремитентно протичане, подобно на МС [6, 31].
От 2006 година съществуват ревизирани критерии за ОНМ, след първоначално предложените през 1999 г. [31, 32, 35] (Таблица 1.). Понастоящем съществуват национални организации за диагноза, лечение и регистриране на случаите с ОНМ (напр. NEMOS Study Group, Germany; www.nemos-net.de; или UK NMO-website www.nmouk.nhs.uk). Лечението на ОНМ, което променя хода на болестта, се базира върху резултатите от ретроспективни наблюдения и консенсусни мнения [4, 6, 7, 12, 25]. Пристъпите при ОНМ са тежки и често водят до траен дефицит, ако не се лекуват адекватно [6, 9, 25, 29]. Повечето болни могат да бъдат в ремисия чрез прилагане на продължителна и адекватна имуносупресивна терапия [6, 9, 12, 25, 27, 26, 29, 34]. Най-удачните терапевтични режими за болните с ОНМ са обект на бъдещи клинични доказателства.

Цел на настоящата статия е да се представят собствени клинични, невроизобразителни, серологични и ликворологични данни при пациенти със сигурна и вероятна диагноза ОНМ и да се дискутират особеностите при тяхната диагноза и лечение.
Контингент и методи
Девет 9 болни (8 жени и 1 мъж на средна възраст 38.0+12.7 г.) са изследвани и лекувани в клиниката по нервни болести при УМБАЛ „Царица Йоанна – ИСУЛ“ София. Диагнозата е потвърдена въз основа на ревизираните критерии на Wingerchuk от 2006 за ОНМ [37]. Пациентите са изследвани клинично (невростатус и EDSS дефицит), лабораторно (ПКК, биохимия, електролити, коагулация, CRP), ликворологично (общо изследване и наличие на олигоклоналност), и относно невроинфекции (HIV, луес, невроборелиоза, VHS, VZV, EBV, CMV). МРТ на главен мозък и цервикален гръбначен мозък е проведена на различни апарати, с 1.5 Т сила на магнитното поле, при различни протоколи. Диагностичните, клинични и образни критерии за ОНМ при различните пристъпи са оценявани ретроспективно. Антителата към AQP-4 в серум и ликвор при 8 от болните са изпращани за изследване в лабораторията по невроимунология към Хайделбергския университет, Германия. Използвана е методиката Cell Based Assay (huAQP4-transf. HEK293-Zellen) [14-16]. При една от болните, наличието на серумни AQP-4 антитела е установено в Испания.
Резултати
Изследваните са разпределени в две групи: (1-ва) 6 болни (от които 1 мъж) със сигурна диагноза (СД) и серопозитивни и (2-ра) 3 пациенти с вероятна диагноза (ВД) при серонегативен статус (две от тях са имали диагноза МС и една – идиопатичен демиелинизиращ процес със при стволова локализация, непокриващ критериите за МС или за ОНМ (таблица 2).
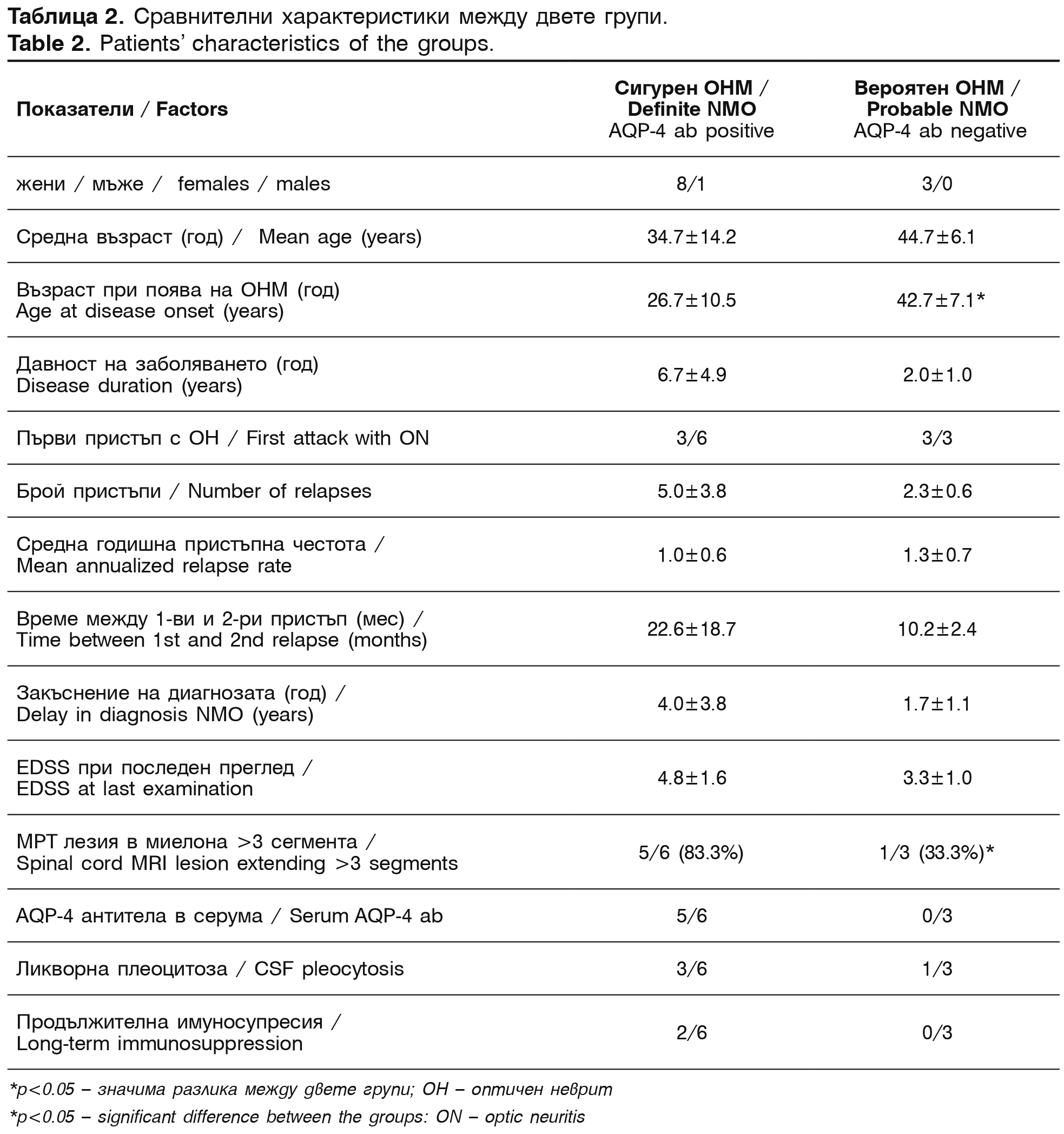
Първият клиничен епизод в групата със СД е започнал с ОН (или ретробулбарен неврит) при 3 от 6 случаи (50%), едностранен ОН при 2, билатерален при 1. Миелит е възникнал при 3 от 6 (50%), едновременно засягане с ОН и М има при 2 (33.3%). В групата с ВД 2 болни имат едностранен ОН, а една двустранен ОН като дебютна симптоматика. Всички болни са имали рецидивиращ ход на заболяването. При средно проследяване от 6.7 години (диапазон 1-14) болните със СД са имали средно 5+3 рецидива и средната годишна пристъпна честота 1.0+0.6 (диапазон 0.4-2.0). Средният интервал между първи и втори пристъп при серопозитивните болни с ОНМ е 22.6+18.7 месеца (диапазон 1-52). При 5 от 6 от случаите (83%) втората атака е възникнала в рамките на 2 години, а при 1 болна (17%) до 5-та година. Средата инвалидност според EDSS при последния пристъп на болните със СД e 4.8+1.6 срещу 3.3+1.0 за втора група. Резидуалният дефицит по EDSS е нараствал прогресивно след всеки нов пристъп в първата група.
Серумните проби за наличие на AQP-4 антитела при общо 4 от всички 9 болни (44.6%) са отчетени качествено като положителни, при чувствителност 78% и специфичност 100% [15]. Една от пациентките със СД не е изследвана серологично, поради невъзможност да се изпрати проба. При друга една болна са установени AQP-4 антитела в серума в лаборатория в Испания (не може да се посочи специфичност и сензитивност). Паралелни серуми и ликворни проби са изследвани при 2 от 9 болни (22%), като при една болна със СД са установени също AQP-4 IgG антитела и в ликвора. Всички болни с ВД имат негативни серумни проби за AQP-4 антитела и отрицателна ликворна проба при 1 изследвана.
Стандартно ликворно изследване е проведено при 7 от 9 болни (71.4%, 5 болни със СД и 2-ма с ВД). Най-честата находка е била лека плеоцитоза, под 50.106/л лимфоцити (3 от първа група) и умерена над 100.106/л лимфоцити (1 от първа група). Данни за олигоклоналност не са установени (0/5), а повишен общ белтък под 0.6 г/л е имало при 3 от 5 изследвани от първа група.
Типични МРТ находки за ОНМ са установени при всички болни със СД освен при една, която няма МРТ изследване на гръбначен мозък. Лонгитудинална демиелинизираща лезия в цервикалния миелон, разположена на три или повече сегмента се открива при 5 от 6 болни (83%) със СД (Фигура 1) и само при 1 с ВД (33%). Атипична голяма МРТ лезия в мозъчния ствол е намерена при 1 болна от втората група, на границата понс – медула облонгата. Данни за торакален миелит са установени при 4 от 6 (75%) от първата група и при 1 от втора. Нормална МРТ или неспецифични малки демиелинизиращи лезии в главния мозък (перивентрикуларно или в ствола и малкия мозък) са открити при всички болни със СД и при 2 с ВД.
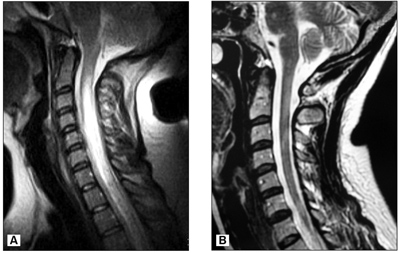
Фигура 1. Характерна Т2-хиперинтензна надлъжна лезия в цервикалния миелон при 27-годишна болна с ОНМ, разположена от С1 до С8 сегмент (A). Отделни Т2-хиперинтензни лезии при болна с клинично сигурна МС, разположени на нивото на С1, С2-3 и С5-6 сегмент (B).
Придружаващи системни заболявания и лабораторни патологични резултати са регистрирани при 4 от 6 болни от първа група и при 1 от втора (33%): алергичен ринит, невроувеит, температура, бронхопневмония или инфекциозно заболяване до 4 седмици преди поява на пристъп на ОНМ, наличие на ANA и АPL антитела (1/6), автоимунен тироидит (0%), положителни HCV антитела (0%), епилепсия (0%), миастения гравис (0%) и др.
Всички пациенти са лекувани с кортикостероиди венозно (най-често 6-метилпреднизолон в дози 250-1000 мг) по време на пристъпите. Допълнително имуносупресивно лечение с азaтиоприн е приложено при 2 болни от първа група (след 8-ми и съответно 4-ти пристъп), и при 1 болна (след 2-ра атака) от втората група. Имуномодулатори не е получавал нито един от болните. Имуноглобулини (IgG, имуновенин) са прилагани периодично при 1 пациент след 4-тия пристъп, в доза 0.2 г/кг месечно, в продължение на 3 години, като е отчетено стабилизиране на статуса, при персистиране на зрителния и гръбначномозъчен остатъчен дефицит. При този болен е проведена и еднократна плазмофереза на десетата година от заболяването, след идентификацията на AQP-4 антитела. При една от болните е поставен вентрикулен шънт, поради МРТ данни за повишено интракраниално налягане.
Само един серопозитивен пациент с ОНМ от всички описани е починал (1/6, 16.6%). Непосредствени причини за смъртта при този болен са епилептичен статус, хипонатриемия, пневмония и мозъчен оток.
Обсъждане
ОНМ е специфично демиелинизиращо заболяване на ЦНС, което протича с тежко засягане на зрителните нерви и миелона. Това заболяване понякога трудно се различава от МС, особено в ранния стадий или при първи пристъп. НМО обикновено се характеризира с пристъпен ход. При симултантна поява на ОН и М, заболяването трудно може да се диференцира и от остър дисеминиран енцефаломиелит. Около 30% от пациентите с ОНМ умират в първите 5 години, 50% от останалите имат тежко увреждане на зрението и не могат да се придвижват самостоятелно [7, 12, 27, 29-31]. Клиничното прогресиране на това В-клетъчно медиирано заболяване е в резултат на уврежданията, които са свързани с инфилтрация на макрофаги, гранулоцити и еозинофили, отлагане на IgM и комплемент [14, 20]. Демиелинизацията и кавитацията на зрителните нерви и миелона, некрозата на сивото и бяло вещество, съдовата хиалинизация и фиброзата, астроцитната некроза са сред най-важните аспекти в патогенезата на ОНМ [33, 35].
Спектърът на заболяванията с AQP-4 антитела в серума, освен класическия ОНМ, включва парциални форми на невромиелит – рекурентен изолиран ОН, остър трансверзален миелит, рекурентен миелит, билатерален ОН, ОНМ със стволова лезия (АДЕМ/AQP4-регион), педиатричен ОНМ, мозъчностволов ОНМ (при хипоталамични или периакведуктални лезии, злокачествено хълцане или повръщане), миастения гравис AQP-4 антитела, ОНМ асоцииран със системни болести (системен лупус, синдром на Sjögren) или азиатска оптикоспинална МС [3, 24, 33].
Нашето първо пилотно за България изследване представя малка група болни с ОНМ, които имат сходни характеристики с класическите описания [3, 7, 9, 11, 30, 35]. Засегнати са предимно жени в 3-то десетилетие, като средната възраст при дебюта на ОНМ е 26.7+10 г (само една болна е била на 47 г.). Според други автори, началото на ОНМ обикновено е около 40 години [4, 10, 29, 34, 35]. Диагнозата ОНМ отговаря на съвременните ревизирани критерии при всички болни от първа група. Само при една болна от тази група няма серологично изследване за AQP-4 антитела, но нейната МРТ с типична цервикална увреда и клинична презентация с оптичен неврит, и последвали още два епизода на миелит отговарят на останалите критерии на Wangerchuk, 2006. Поради тежкия трансверзален миелит в шийната интумесценция, при тази болна клинично са наблюдавани тонични болезнени спазми, както се описва при някои случаи с ОНМ [17, 24, 27]. Тези инвалидизиращи симптоми при нашата болна преминават за няколко седмици след пулсова терапия с кортикостероиди и симптоматично приложение на тизанидин и габапентин в големи дози. При един от пациентите са наблюдавани двукратно пристъпи на злокачествено хълцане и повръщане (на 2-та и 7-та година от заболяването), което е специфичен синдром, описван в литературата за ОНМ [10, 25-27, 31].
Изследването на главния мозък и миелона чрез МРТ при 85-100% от нашите случаи потвърждава резултатите от други автори относно наличие на малко неспецифични супртенториални лезии и на една лонгитудинална и централно разположена лезия в миелона, над 3 сегмента [5, 10, 27, 35]. При 2 болни от първа група (33.3%) гръбначният мозък е засегнат на две и повече места. Възможно е миелонът да бъде интактен при малка част от болните с ОНМ [8, 11, 19], както ние установихме при 2 болни от втора група. Мозъчните лезии при ОНМ могат да бъдат специфични или неспецифични [3, 5, 16, 19, 24, 26]. Смята се, че при изследване с МРТ 10% от болните с ОНМ изпълняват критериите на Barkhof за МС. Доскоро не само за нашата страна, но и по света много от болните с ОНМ са диагностицирани като вариант на МС [2, 9, 10, 12, 18, 21, 24, 30]. Най-често лезиите в главния мозък при ОНМ са в зоните с висока експресия на аквапoриновите канали в ЦНС [26]: хипоталамуса, дианцефалона, около четвърти вентрикул, корпус калозум (с линеарно засягане), но понякога лезиите са огромни (т. нар. „tumefactive lesion”) [19].
При трите болни с ВД ОНМ не се установиха AQP-4 антитела в серума (възможност за фалшиво негативни резултат под 2%), въпреки че при тях заболяването започва с оптичен неврит (двустранен при една пациентка). Само една от тях има засягане на миелона на три сегмента (Т6-Т9), докато при другите две липсва диагностичния критерий – лонгитудинална лезия в миелона. При една от тези болни е наблюдавана типична стволова симптоматика (с тежък дискоординационен, квадрипирамиден и алтерниращ сетивен синдром). При нея възниква парамедианна демиелинизираща увреда в зоната на понса и медула облонгата, една година след двустранен ретробулбарен неврит, с остатъчна амблиопия. Изолирано засягане на мозъчния ствол, а не на цервикалния миелон при ОНМ е рядко, но възможно представяне [8, 12, 27, 29, 33, 35]. Това беше основание да изследваме нейната серумна и ликворна проба за наличие на AQP-4 антитела.
Промените в ликвора [6, 11, 22, 23] показват обикновено плеоцитоза над 50.106/л при 13–35% от болните (само при 3 наши болни), а при отделни случаи до 1000.106/л (1 от нашите пациенти) [22, 30, 35]. Повишени стойности на общ белтък се установяват при двама от нашите случаи. Олигоклоналност при ОНМ е описана при от 1% до 37% [7, 22, 23]. Такава липсва при нашите болни. При установяване на олигоклоналност и 1–2 мозъчни неспецифични МРТ лезии, диагнозата ОНМ не се изключва, но е необходимо клинично и МРТ проследяване. В обратните случаи диагнозата МС е по-вероятна.
В сравнение с МС, средната пристъпна честота е по-висока при болните с ОНМ (1.3 срещу 0.6) [34], както наблюдавахме и ние. Честотата на втория рецидив в рамките на първите две години (82% при ОНМ срещу 62% при МС) [34]. Средното време за достигане на EDSS 3.0 и 6.0 също е по-кратко при ОНМ (съответно 0.5 и 7 г.) отколкото при МС. За идентифициране на рисковите фактори, прогнозиращи хода и преживяемостта при ОНМ, Wingerchuk и Weinshenker [2003] доказват, че прогнозата е по-добра при болни с начало на ОНМ преди 40 години, с по-лек дефицит след изява на М, както и с по-дълъг интервал между първия и втория пристъп, и при тези с ниска честота на рецидивиране.
Смята се, че смърт възниква само при рецидивиращия ОНМ и e свързана с наличие на автоимунно заболяване, степента на моторно възстановяване след М и броя на екзацербациите през първите две години от началото [4, 34]. Само един от нашите пациенти със серопозитивен ОНМ е починал (1/6, 16,6%) след десет годишна еволюция на заболяването (при над 12 рецидива, няколкогодишно стабилизиране от интермитентно приложение на имуновенин). Шест месеца преди това при него е осъществена една процедура на плазмообмяна, а до момента на последната хоспитализация, той е приемал азатиоприн и малки дози преднизолон.
Важно е да се отбележи, че нито един от нашите пациенти до установяване на AQP-4 антителата не е лекуван с продължителна перорална имуносупресия или с интермитентна плазмафереза.
Средното закъснение от първия пристъп до поставяне на правилната диагноза ОНМ при нашите 6 болни от първа група е 4.0+3.8 години. Поради голямата давност на заболяването и множество рецидиви при двама от болните, това закъснение е до 8-9 години. Други диагнози, които са описвани през годините при нашите пациенти, (преди окончателно да се приеме ОНМ), са били: ОН, ретробулбарен неврит, рецидивиращ неврит и М, трансверзален М, пристъпно-ремитентна, хронично проградиентна и предимно спинална форма МС, цервикална миелопатия, интрамедуларен тумор, сиригомиелия, нормотензивна хидроцефалия, енцефаломиело-полирадикулоневрит, световъртеж от централен произход, клинично изолиран синдром, хроничен демиелинизиращ процес, антифосфолипиден синдром, синдром на Девик, кома неиндуцирана от травма и др. Диагноза МС предварително са имали 3 (50%) от онези 6 болни със сигурен ОНМ.
Като някои причини за късно поставяне на диагнозата ОНМ при нашия контингент могат да се посочат: липса на обзорни статии за болестта на български език (2), непознаването на критериите нa Wingerchuk преди 2006 г., невъзможност за изследване на специфичните антитела, извършване на МРТ само на главен мозък при някои болни с ОН, но не едновременно и на миелона, приемане на рецидивите на заболяването за рецидиви на МС, повлияването на симптомите при някои болни в краткосрочен план след приложение на КС, и/или липсата на системно проследяване на болните от един специалист.
В заключение, пациентите с доказани AQP-4 антитела отговарят на всички съвременни критерии за ОНМ. Сравнението на данните между двете групи е относително, поради малкия брой изследвани, но беше необходимо за апробация на критериите. Най-голямо клинично значение за поставяне на диагноза ОНМ има стриктното познаване и прилагане на международните критерии. Тази самостоятелна болестна единица трябва да се разграничава рано от МС и други демиелинизиращи заболявания. Тя винаги трябва да се търси при болни с рецидивиращ ОН и М, когато МРТ находката на главен мозък е нормална и липсват олигоклонални фракции в ликвора. Необходимо е натрупване на информация за повече болни и създаване на регистър за ОНМ в България.